It’s been said a million times before but it’s worth re-emphasizing.
Creating.
a.
new.
drug.
is.
hard.
With so many steps in the drug discovery process, the room for failure is massive. But it doesn’t always have to be that way. At Deep Origin, we’ve been working on building computational models that are more predictive, increasing the chances of success in drug development. Here we describe our work on building a better pocket-finder model, which is used for our docking workflow in our AI assistant Balto.
Molecular docking is a computational method that predicts how a small molecule (a potential drug) will bind to a larger biological molecule (the target, like a protein) by simulating their interaction, determining the best position, orientation, and shape for binding1. Docking models can be categorized by where they dock a ligand - either globally (searching all over the protein) or locally (into a specifically-defined protein pocket). Recent diffusion models like AlphaFold 3 and Boltz-2 fall in the first category. They are strong in predicting overall structure and for working with familiar ligands; however tend to perform worse than models or tools that dock ligands locally when it comes to pose prediction, binder/non-binder classification, and discriminating false positives2. False positives, or molecules that look like they will bind in docking but fail to bind in lab assays, is a loss of both time and money in a drug discovery program. It’s also worth noting that this pocket-based approach is specific to small-molecule drug discovery, as larger modalities like peptides, proteins, and antibodies often engage targets through broader or conformational surfaces rather than well-defined pockets.
To dock small-molecule ligands locally, you need to know where on the protein to dock; this is where pocket-finding algorithms come in. Pocket-finding, usually an early step in docking ligands or small molecules to a target protein, identifies potential binding sites or “pockets” within a protein structure. These pockets are typically cavities or depressions on the protein surface where ligands, such as small molecules or drugs, might bind. Importantly, these pockets typically need to be biologically relevant to the specific drug discovery campaign, such as active sites of enzymes, or pockets at protein-protein or protein-nucleic acid interfaces, since most inhibitor or activator design efforts target functionally meaningful regions on the target protein. Pocket finding algorithms look at a combination of properties, including size and shape of a cavity, hydrophobicity, and potential to form different bond types, to score and rank pockets on a protein. Identifying these pockets is crucial for understanding protein function as well as for rational drug design, as it helps researchers target specific areas on the protein where therapeutic compounds could interact effectively.
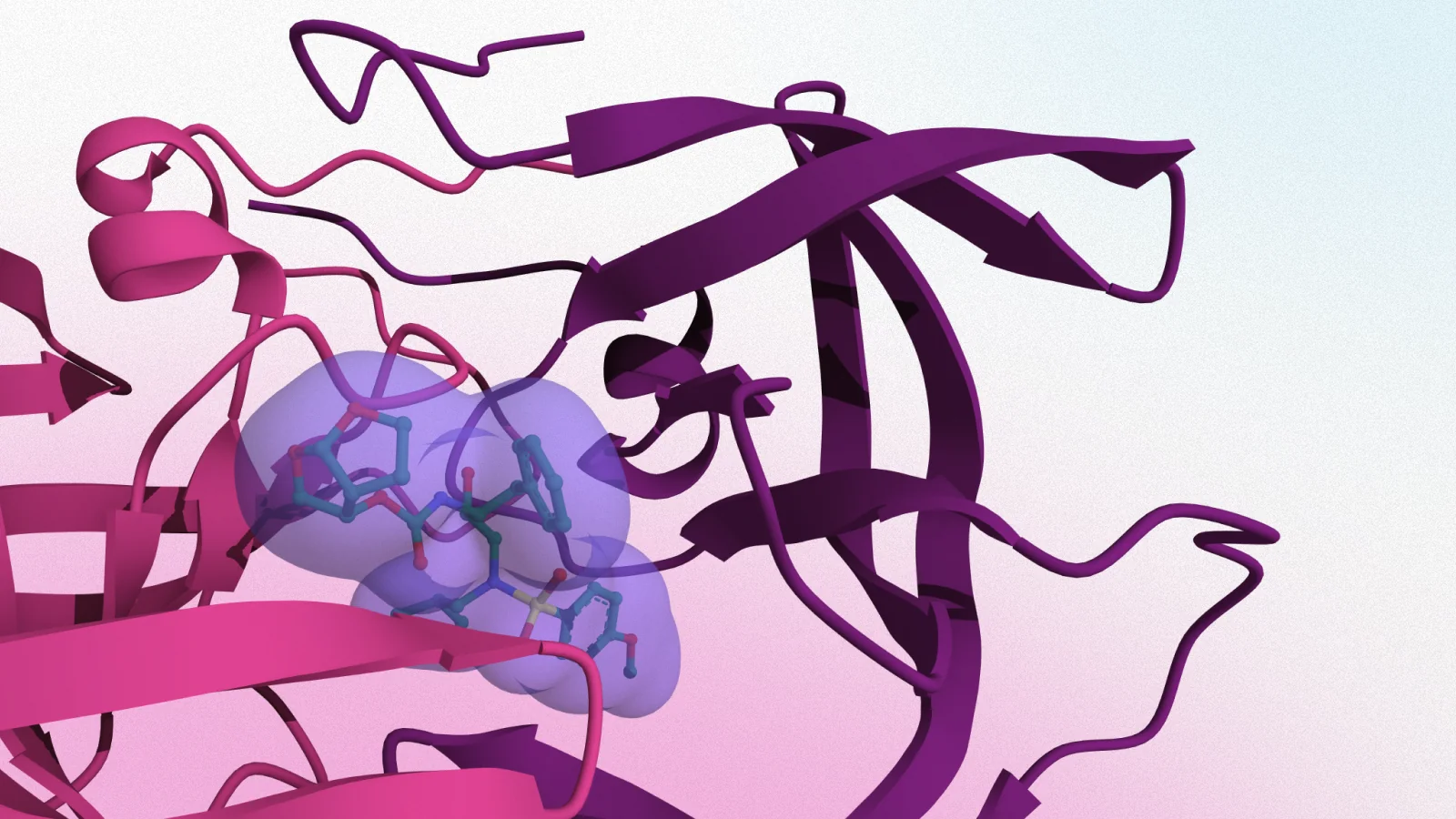
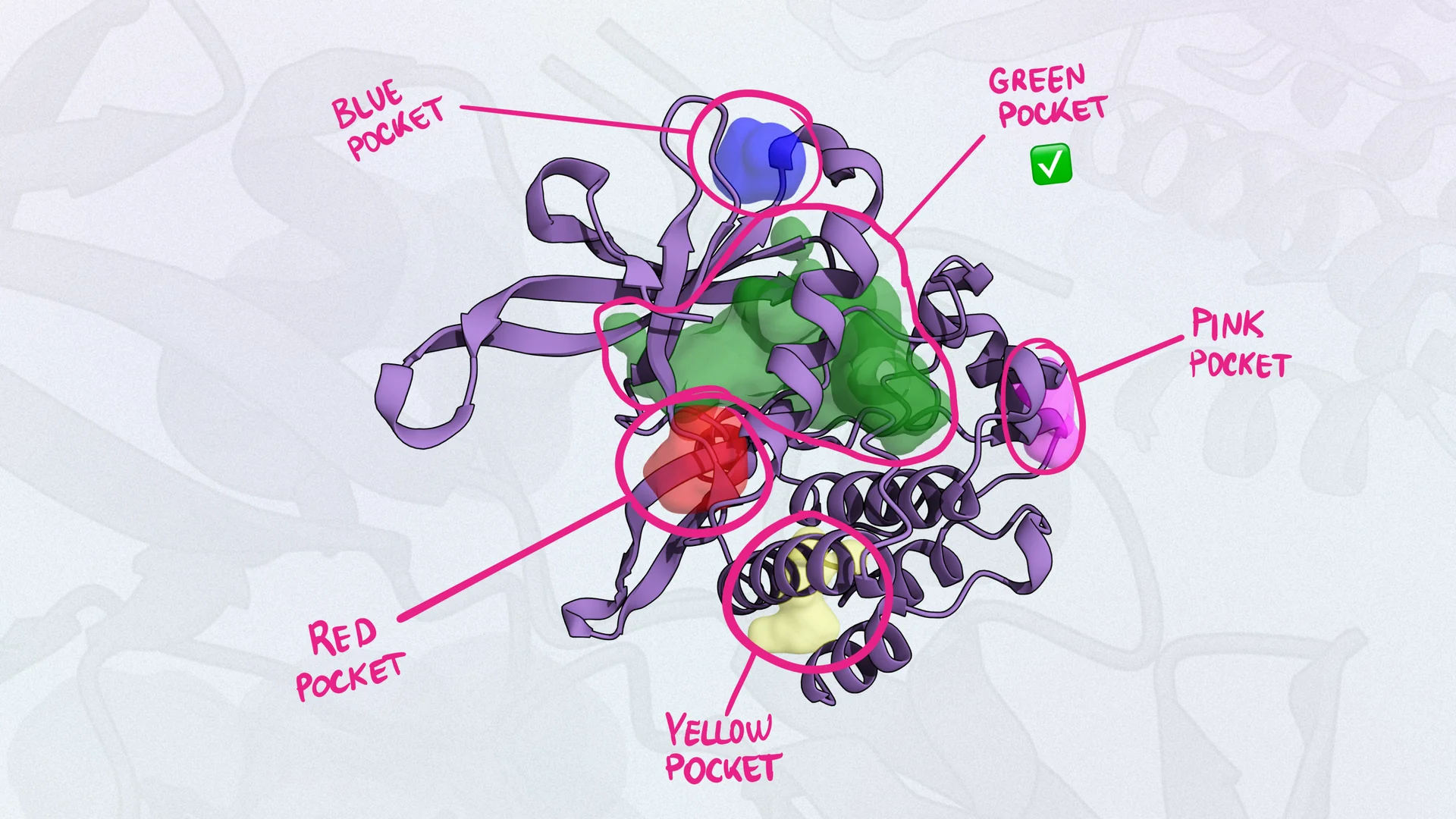
With Balto, you can identify not only the crystal ligand pocket, but also all other potential binding pockets (Figure 1, 2).
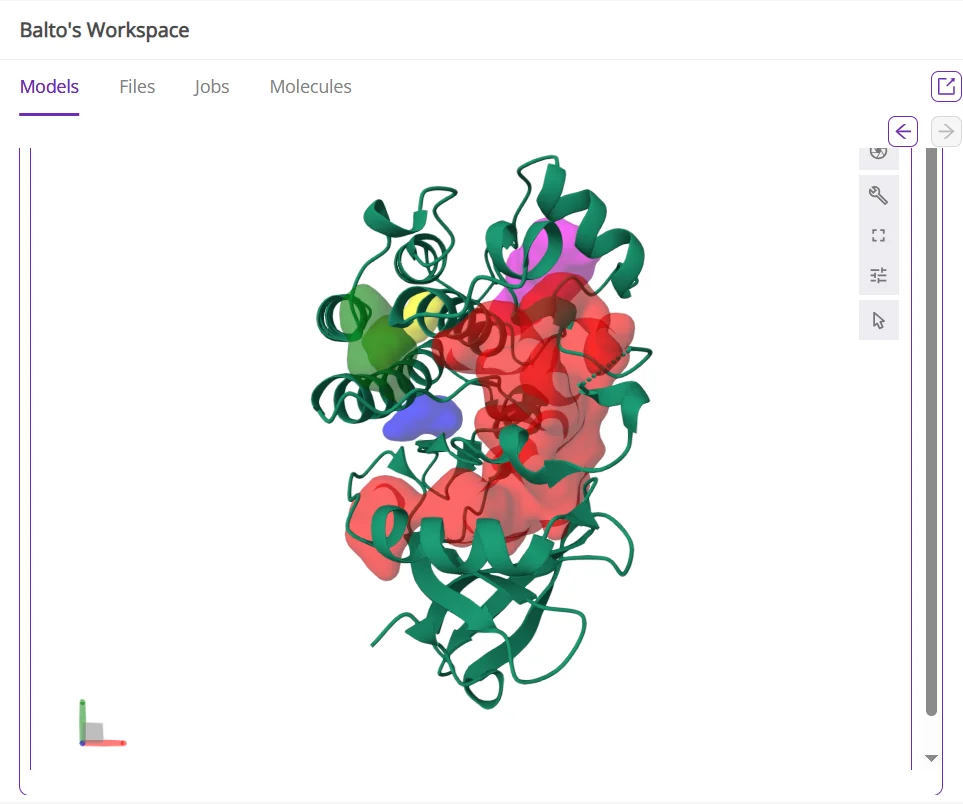
Figure 1: Example of Balto identifying binding pockets in BTK protein (green). The red pocket is the one containing the crystal ligand (ibrutinib).
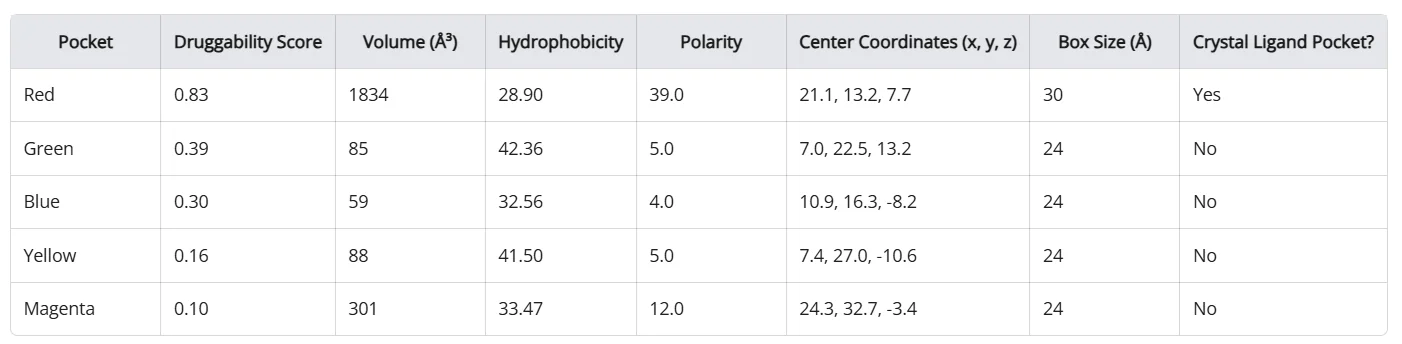
*Figure 2: Table of all pockets Balto identified in the BTK protein. *
However, pocket finding in proteins faces several notable challenges: false positives and hidden or cryptic sites. One of the biggest issues is the prevalence of false positives—situations where algorithms incorrectly identify a region as a binding pocket due to superficial geometrical characteristics rather than genuine binding potential. These inaccuracies can lead to wasted efforts in drug discovery by having scientists target non-viable sites. Additionally, hidden sites pose a substantial challenge, as some potential binding regions are concealed due to the protein’s conformational dynamics and are not easily detectable using static structures alone. This dynamic nature of proteins means that pockets can be transient, appearing and disappearing depending on the conformational state, further complicating accurate identification. Lack of accuracy stems from limitations in current computational methods which may not fully account for the complex interplay of electrostatic and hydrophobic interactions that characterize true binding sites. We’re not quite there yet tech-wise—but we’re continuing to work on it behind the scenes.
Our team was frustrated with all of these and so we set out to improve on the current pocket-finding algorithms. The result is a pocket-finding algorithm with higher accuracy built directly into Balto. While a search for cryptic pockets is not available in Balto, reach out to us for partnerships if you’re interested in that!
More accurate results: the benchmark details
We benchmarked our PocketFinder using Coach420 (Mlig-merged) against GrASP and P2Rank. GrASP (Graph-based Surface Pockets) is a geometry-based pocket detection method established by Santana et al. in 2020 that leverages graph theory to analyze protein surfaces. P2Rank is a machine learning-based ligand-binding site predictor developed by Krivák et al. in 2018, trained on known protein-ligand complexes.
Coach420 is a standard benchmarking dataset used to evaluate protein binding site (pocket) prediction algorithms. It contains 420 proteins with known ligand-bound structures. Some proteins may also have multiple copies of the same ligand (e.g., symmetric sites) or different ligands that bind overlapping or adjacent sites. To account for this, the benchmark was performed under multi-ligand merged format. Multiple ligands per protein were clustered and merged into unified binding sites for a more fair and biologically relevant pocket prediction evaluation.
These results are determined based on Top “N” metrics: Among the top-N predicted pockets for each protein, how many known binding sites are recovered. For example, if a protein has 3 real binding sites, and the model predicts 5 pockets, Top-N Recall checks how many real sites are hit among the top 3 or top 5 predictions. Hence, in 80.60% of cases using DO PocketFinder, at least one of the top 3 predicted pockets matched a real known binding site. While our tool offers only a modest improvement over GrASP in raw accuracy, even a few extra percentage points can significantly reduce false positives, leading to more reliable docking and better experimental prioritization.
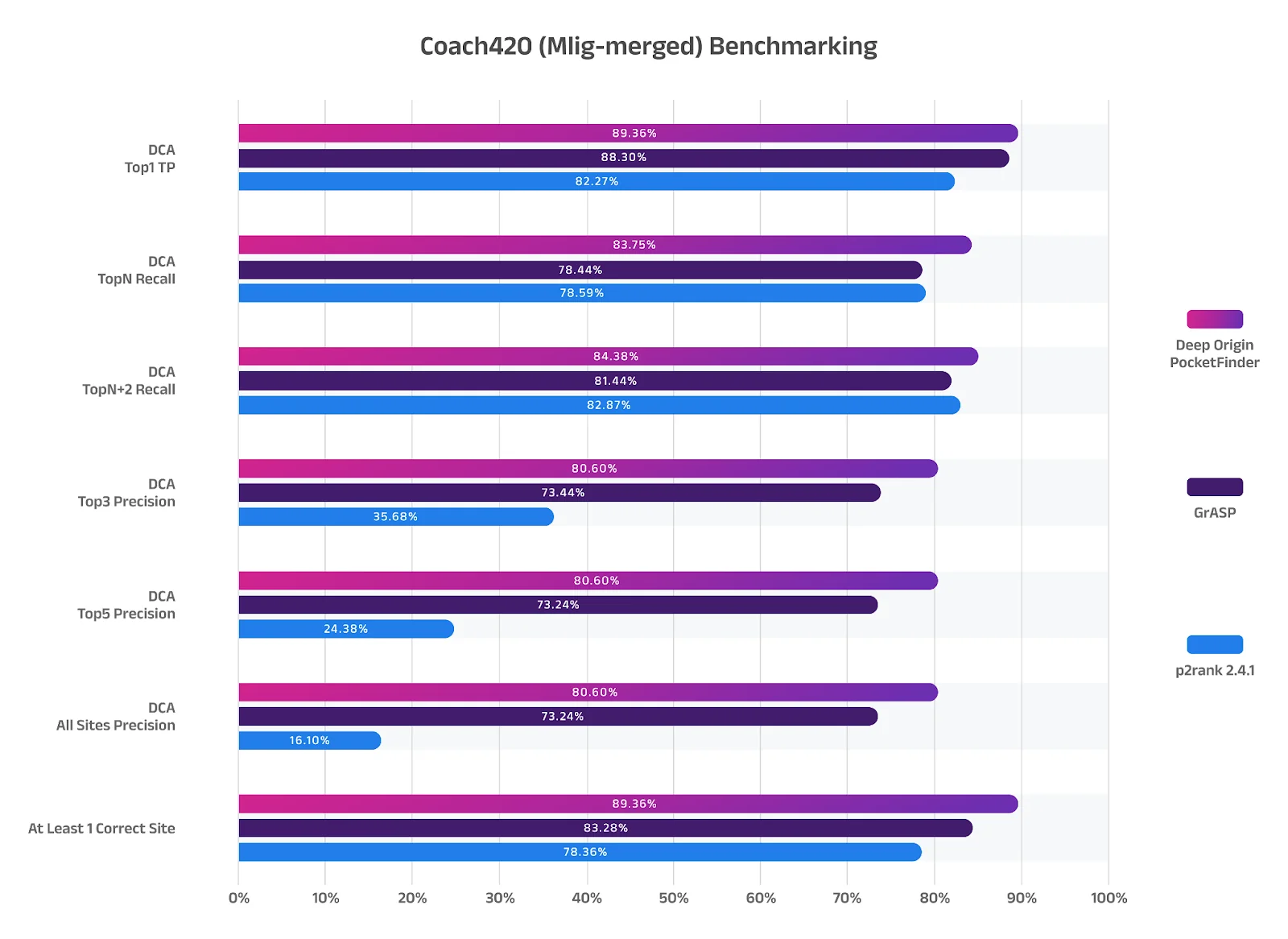
Figure 3: Deep Origin PocketFinder outperforms GrASP and p2rank benchmarking. The same can be interpreted for the other metrics:
- Top1 TP (Top Pocket): How frequently a binding site is the #1 prediction.
- TopN+2 recall: Similar to Top-N Recall, but allows 2 extra predictions per protein.
- Top 3: Of the top 3 predicted pockets, how many are true binding sites?
- Top 5: Of the top 5 predicted pockets, how many are true binding sites?
- All sites: Among all predicted pockets by a method, how many correspond to true binding sites?
- At least one correct site: Did the method predict at least one true binding site for each protein?
In all metrics, our PocketFinder outperformed the benchmarks, showcasing a remarkable >80% score. In other words, >80% of the time our PocketFinder’s top predictions include a real binding site. For most drughunters, the first metric (Top 1) is the most significant. Based on benchmarking results, we’re able to predict almost 90% of actual binding sites.
See it in action!
References
1. Torres, P. H. M. et al. Key topics in molecular docking for drug design. Int. J. Mol. Sci. 20, 4574 (2019).
2. Buttenschoen, M., Morris, G. M. & Deane, C. M. PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chem. Sci. 15, 3130-3139 (2024).