Disclaimer: We tested the publicly available AlphaFold 3 instance and have reviewed the paper. Note that access to the full tool is not yet available and is provided only as a server with limitations. Here’s what we found.
AlphaFold 3, which is the next step of AlphaFold 2, models proteins with possibly modified residues, nucleic acids, small molecules and ions. It uses a single ML model for predictions from input of biopolymer sequences, residue modifications, and ligand SMILES, claiming improved accuracy over specialized tools. Below we’ll work through an internal, quick review of the announcement paper, pointing our noteworthy advances and some significant shortcomings of the next generation of AlphaFold.
In our review of the paper, we noticed some interesting choices made by AF3’s authors. Notably, the paper refers to Autodock Vina as a state-of-the-art (SOTA) docking tool (indeed, it is a great, widely used tool, but far from being SOTA). Also, the paper refers to AlphaFold-Multimer as a highly accurate algorithm. This is quite surprising given the serious accuracy issues that the model is known for.
AlphaFold 3 Architecture
AlphaFold 3 introduces two major architectural changes:
- The EvoFormer module of AlphaFold 2 has been replaced with a simpler Pairformer module to reduce MSA processing. This means MSA construction is not removed from AlphaFold 3.
- The Structure Module that was used to predict 3D structures of proteins has been replaced by a Diffusion module that predicts raw atom coordinates.
Interestingly, the Diffusion module operates directly on 3D coordinates instead of torsions, translations, and rotations, where the latter is the approach of other diffusion-based models.
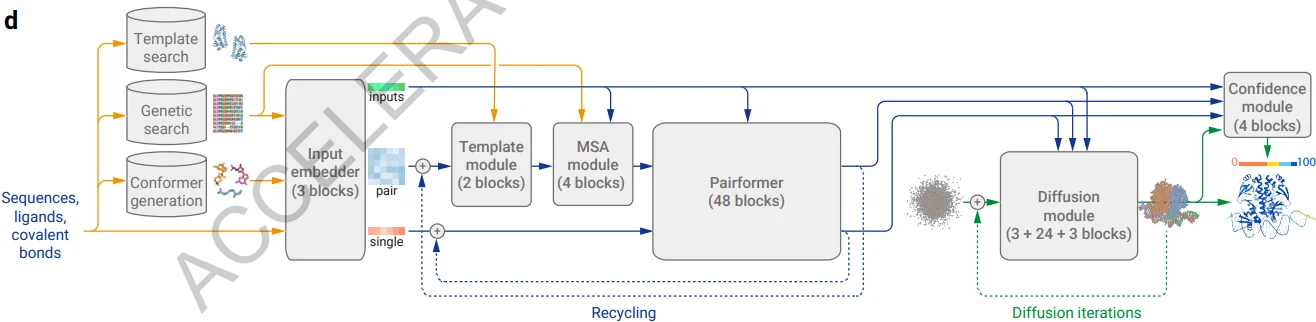
AlphaFold 3 uses cross-distillation with AlphaFold-Multimer v2.3 and AlphaFold 2 to decrease hallucination caused by Diffusion. As in AlphaFold2, confidence measures are used to assess the atom-level and pairwise errors in final structures and are used to select the high-quality structures from sampled candidates.
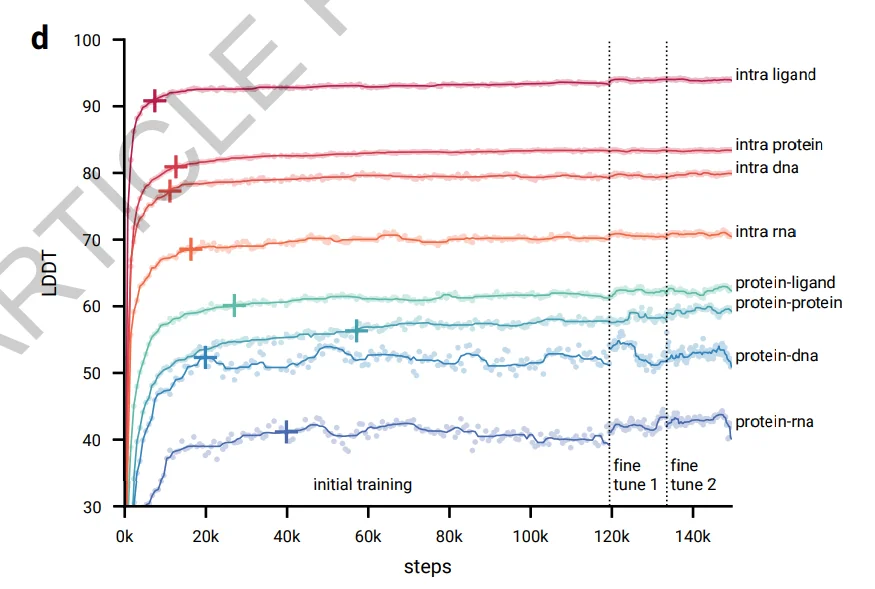
From Fig. 2d it is obvious that the model learns local structures relatively fast in only 20K steps. Interface metrics also achieve high values relatively quickly. This means it’s possible to achieve almost the same accuracy in ¼ of the training steps.
For protein-ligand interactions, the paper used PoseBusters benchmark set, composed of 428 protein-ligand structures released to the PDB in 2021 or later, and trained AlphaFold3 using a 2019-09-30 training cutoff. Here we have a time split without homology or ligand similarity splits. This is definitely not the hardest split to use for ML benchmarking. AF3 accuracy on the PoseBusters V1 set is 76.4%, which is 2.8% improvement over their recent technical report. In Extended Data Fig. 3 we can see that when they used a homology filter for proteins on complexes where the protein is not similar to the training set, AF3’s accuracy isn’t, surprisingly, deteriorated.
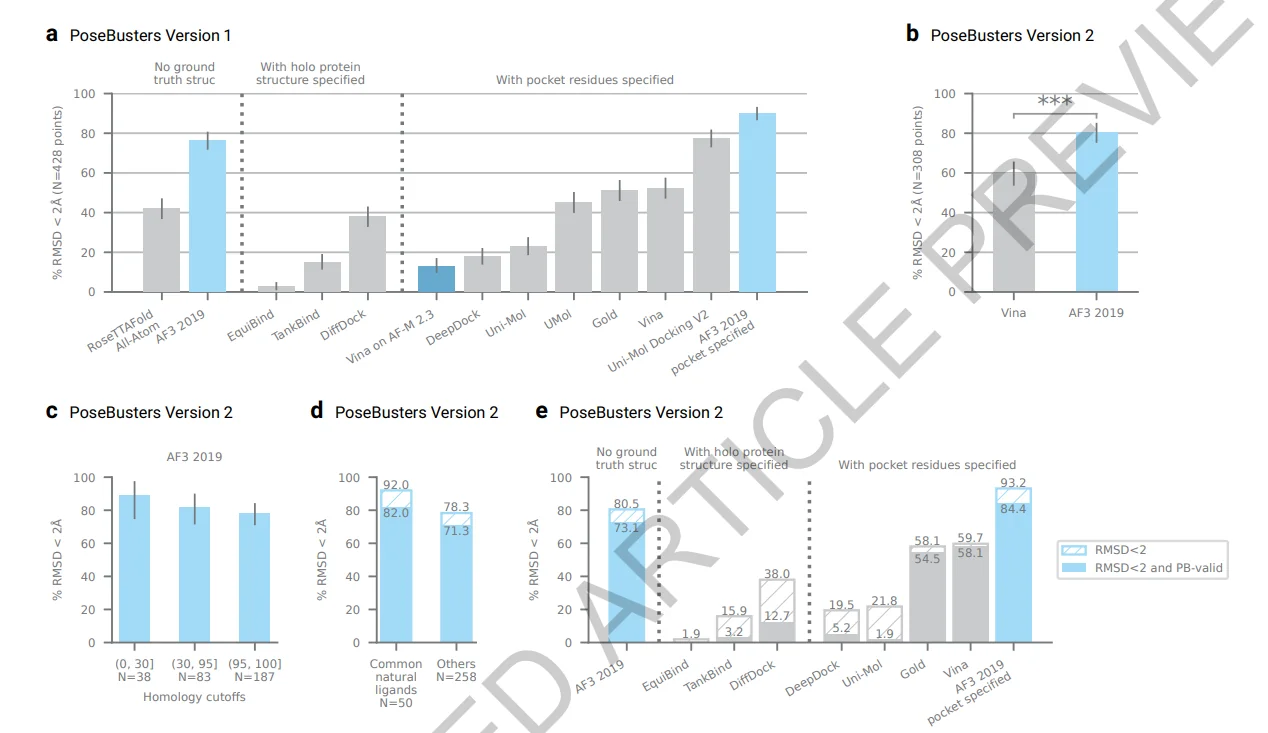
Unfortunately, they don’t show ligand-based splitting as well. This is interesting for two reasons:
- In AlphaFold’s latest paper, we saw that the most challenging split for AF is the ligand-based split (see Figure 5a).The results greatly deteriorated when low similarity ligands were predicted.
- the accuracy difference between Common natural ligands (defined as those which occur greater than 100 times in the PDB and others is more than 10% (see Extended Data Figure 4).
These two points indicate ligand-based memorization. However, the prediction accuracy of the model on novel ligands is critical because in drug discovery projects we usually would like to find novel molecules.
It is also interesting that in their validation set AF3 used harder splits < 40% sequence identity for biopolymers (see Supplementary information 5.8). Interestingly, in their white paper, they used a threshold of < 0.5 to define novel ligands. However, surprisingly, in the published article, a < 0.85 threshold was used in the validation split, making it likely that models overfitting on ligands got selected. It would have been preferable for the validation set to be both low sequence identity and low ligand similarity. Because our discussion here is based on the validation set, it is difficult to make definite conclusions, however, their test set uses a time split. Therefore, their test set presumably highly overlaps with the training set with regard to structural and ligand similarities, making it difficult to assess model’s performance on novel ligands and pockets.
Interestingly, the allowed ligands in the public server include: CCD_ADP, CCD_ATP, CCD_AMP, CCD_GTP, CCD_GDP, CCD_FAD, CCD_NAD, CCD_NAP, CCD_NDP, CCD_HEM, CCD_HEC, CCD_PLM, CCD_OLA, CCD_MYR, CCD_CIT, CCD_CLA, CCD_CHL, CCD_BCL, CCD_BCB. We would have liked that for the public to be given access to arbitrary ligands docking so an independent assessment of AF3’s docking power can be determined.
Finally, the authors benchmark AF3’s accuracy when only the binding site is specified (Fig. 1a). In this case, they train a new version of AF3 by giving an additional token feature specifying pocket-ligand pairs, providing additional information than using only a pocket box. The latter is how programs like Vina are run, which is closer to real world circumstances.
AlphaFold 3 Model Limitations
In the Model limitations section, the authors highlight a few major limitations of AF3 model:
- Model outputs do not always respect chirality despite the model receiving reference structures with correct chirality as input features. They address this in the PoseBusters benchmark, by a penalty for chirality violation in the ranking formula for model predictions. Despite this, there is a chirality violation rate of 4.4% in the benchmark. Another issue is clashing atoms in the predictions which is again targeted by penalty during ranking.
- AlphaFold 3 model introduces the challenge of hallucinations in disordered regions.
- AlphaFold 3 predicts only static structures, while dynamical behaviors of biomolecular systems are not addressed. Even multiple random seeds for either the diffusion head or the overall network do not produce an approximation of the real-world dynamical ensemble. The conformational diversity problem remains.
- In some cases, modeled conformational state may not be correct or comprehensive given the specified ligands and other inputs. AF3 does not handle conformational changes during binding.
It is interesting that almost all of these limitations can be solved by physics based methods, therefore, it seems that AF3 has problems with modeling physics.
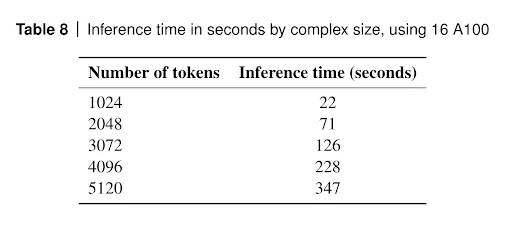
AlphaFold 3 is a large system and uses diffusion iterations. Since the system mainly consists of neural networks, its inference is done on GPUs. It is not a fast method. The GPU wall clock time without MSA construction, data processing and post-processing is given in Supplementary information 5.10 Table 8.
AlphaFold 3 is great. But is it useful?
In conclusion, AlphaFold 3 represents an advancement in the field of biomolecular structure prediction, embodying both notable improvements and significant unresolved problems. Its ability to integrate various molecular components, from proteins to small molecules, using a single machine learning model, may potentially accelerate certain aspects of drug discovery. The architectural changes introduced—such as the replacement of the EvoFormer with the Pairformer, and the switch to a diffusion module for predicting atomic coordinates—demonstrate innovative approaches to enhancing model accuracy and efficiency.
However, despite these advancements, AlphaFold 3 is not without significant limitations. Issues, such as potential prediction deterioration for novel ligands, chirality violations, atom clashes, and the challenge of modeling dynamic biomolecular behaviors suggest that while AF3 enhances certain predictive capabilities, it is far from being a panacea for all the complexities of molecular biology. For example, the well-known extreme sparsity of structural data for RNA and even protein-ligand complexes, raises serious scientific concerns whether an AF3 like model can be truly predictive out-of-distribution. This remains to be seen. On the other hand, the continued reliance on a large overall dataset and substantial GPU resources needed, also implies that widespread accessibility and utility might be constrained. Lack of meaningful public access to AF3 makes its potential impact somewhat limited compared with AF2.