Protein-Ligand Interactions
Computational methods allow for the rational design of ligands that can specifically bind to target proteins through predicted protein-ligand interactions. Virtual screening can evaluate up to billions of compounds *in silico*, identifying potential ligands faster and more cost-effectively than traditional high throughput screening.
Protein-ligand interactions refer to the binding between a protein and a ligand, which can be a small molecule, peptide, or another protein. These interactions are crucial in many biological processes, including enzymatic activity, signal transduction, and cellular communication.
In drug discovery, proteins often serve as targets for therapeutic agents. Ligands (potential drug molecules) bind to specific sites on the protein, modulating its activity. Understanding these interactions helps in designing molecules that can precisely target and influence protein functions, leading to the development of new drugs.
Protein-ligand interactions include Van der Waals (corresponds to shape complementarity), electrostatic (e.g. ionic), hydrogen bonds (distinct directionality and distances), hydrophobic (non-polar ligands escaping water).
Importance in Computational Drug Discovery:
- Rational Drug Design: Computational methods allow for the rational design of ligands that can specifically bind to target proteins through predicted protein-ligand interactions.
- High Throughput Screening: Virtual screening can evaluate up to billions of compounds in silico, identifying potential ligands faster and more cost-effectively than traditional high throughput screening.
- Prediction of Binding Affinity: Computational tools can predict how strongly a ligand binds to a protein, guiding the optimization of lead compounds.
- Understanding Mechanisms: Molecular dynamics simulations and docking studies provide insights into the mechanisms of interaction, aiding in the identification of key binding residues and conformational changes.
Key Tools
-
AutoDock Vina:
-
A popular molecular docking software that predicts how small molecules bind to a receptor of known 3D structure.
-
Schrödinger Suite:
-
A comprehensive suite of tools for molecular modeling, including Glide for docking, Maestro for visualization, and Prime for homology modeling.
-
GROMACS:
-
A molecular dynamics simulation software used to study the physical movements of atoms and molecules, providing insights into the stability and dynamics of protein-ligand complexes.
-
DeepOrigin Tools:
-
Docking: For docking ligands into protein binding sites.
-
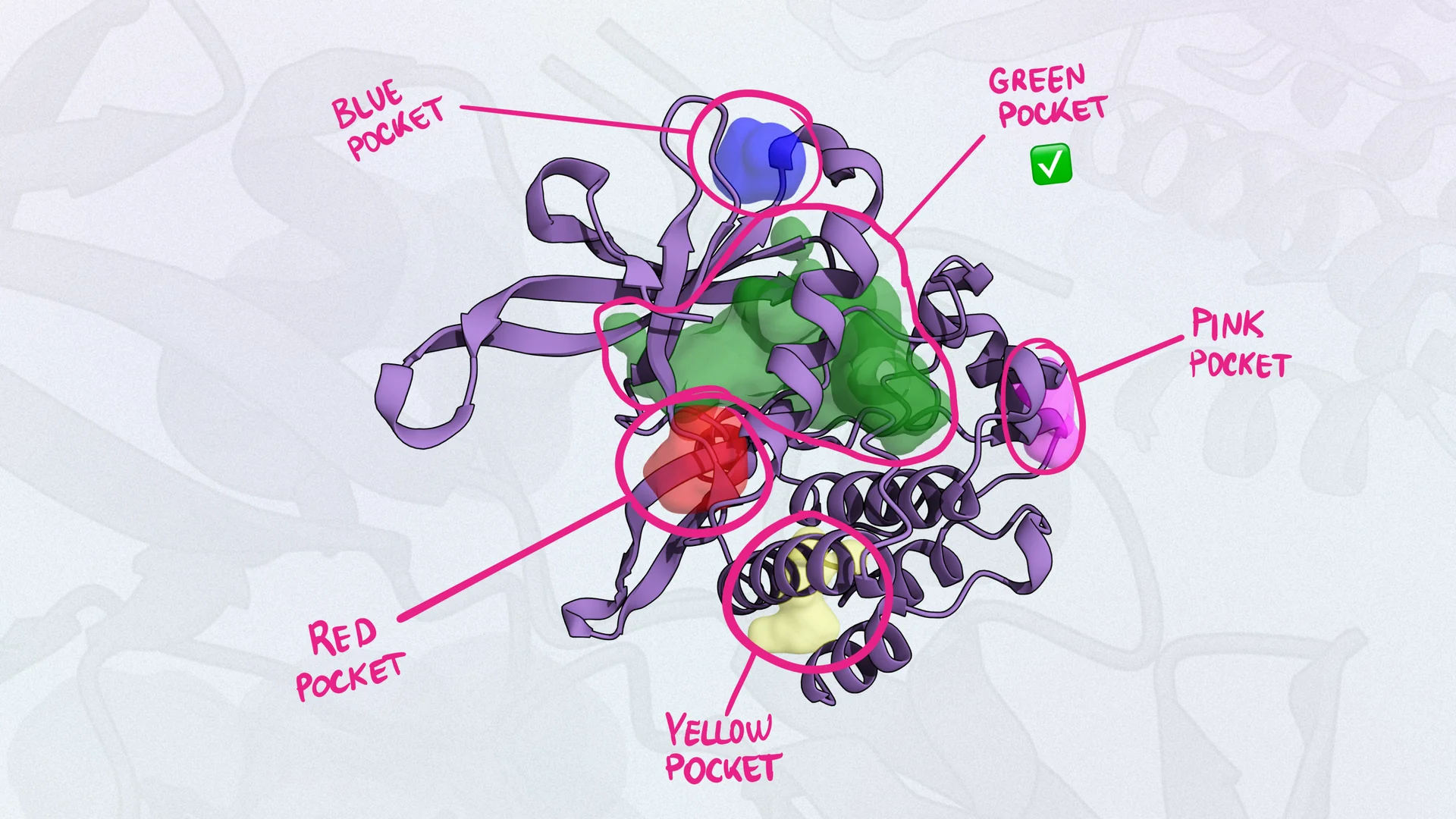
PocketFinder: For identifying potential binding pockets on proteins.
-
BindingDBToolExecutor: For retrieving experimental binding data from databases like BindingDB.
Literature
Computational Docking Analysis of Protein-Ligand Interactions in Diabetes-Related Complications: Targeting Molecular Pathways for Therapeutic Intervention
- Publication Date: 2024-10-01
- DOI: 10.29070/1xcpbp06
- Summary: Focuses on key amino acid residues from proteins involved in diabetes, using various docking methods to investigate binding mechanisms and affinities of potential anti-diabetic medications.
Surface plasmon resonance spectroscopy for characterization of membrane protein-ligand interactions and its potential for drug discovery
- Publication Date: N/A
- DOI: 10.1016/j.bbamem.2013.04.028
- Summary: Describes the use of SPR for characterizing ligand binding interactions with membrane proteins, including major drug targets like GPCRs.
LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery
- Publication Date: 2011-10-05
- DOI: 10.1021/ci200227u
- Summary: Introduces a graphical system for generating multiple 2D diagrams of ligand-protein interactions from 3D coordinates, facilitating analysis tasks in drug discovery.
Protein-Ligand Interactions and Drug Design
- Publication Date: N/A
- DOI: 10.1007/978-1-0716-1209-5
- Summary: Provides an overview of the principles governing protein-ligand interactions and their application in rational drug design, highlighting their role in developing targeted therapeutics.
G protein-coupled receptors (GPCRs): advances in structures, mechanisms and drug discovery
- Publication Date: 2024-04-10
- DOI: 10.1038/s41392-024-01803-6
- Summary: Focuses on GPCR drug discovery, revealing detailed drug-target interactions and underlying mechanisms of orthosteric drugs approved by the FDA.
Building machine learning models to explore protein-ligand interactions for drug discovery
- Publication Date: N/A
- DOI: N/A
- Summary: Discusses the development of machine learning models to predict protein-ligand interactions, aiding the drug discovery process.
An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking
- Publication Date: 2019-03-15
- DOI: 10.1007/s12539-019-00327-w
- Summary: Reviews different types of scoring functions used in molecular docking, discussing their foundations, applications, and challenges.
CHARMM‐GUI high‐throughput simulator for efficient evaluation of protein–ligand interactions with different force fields
- Publication Date: 2022-08-13
- DOI: 10.1002/pro.4413
- Summary: Introduces CHARMM‐GUI HTS, which prepares MD simulation systems for multiple protein–ligand complexes, improving docking results and facilitating hit discovery in drug design.
Mass spectrometry based tools to investigate protein-ligand interactions for drug discovery
- Publication Date: 2012-05-15
- DOI: 10.1039/c2cs35035a
- Summary: Reviews the use of mass spectrometry and related techniques for investigating protein-ligand interactions, highlighting its application in drug development.