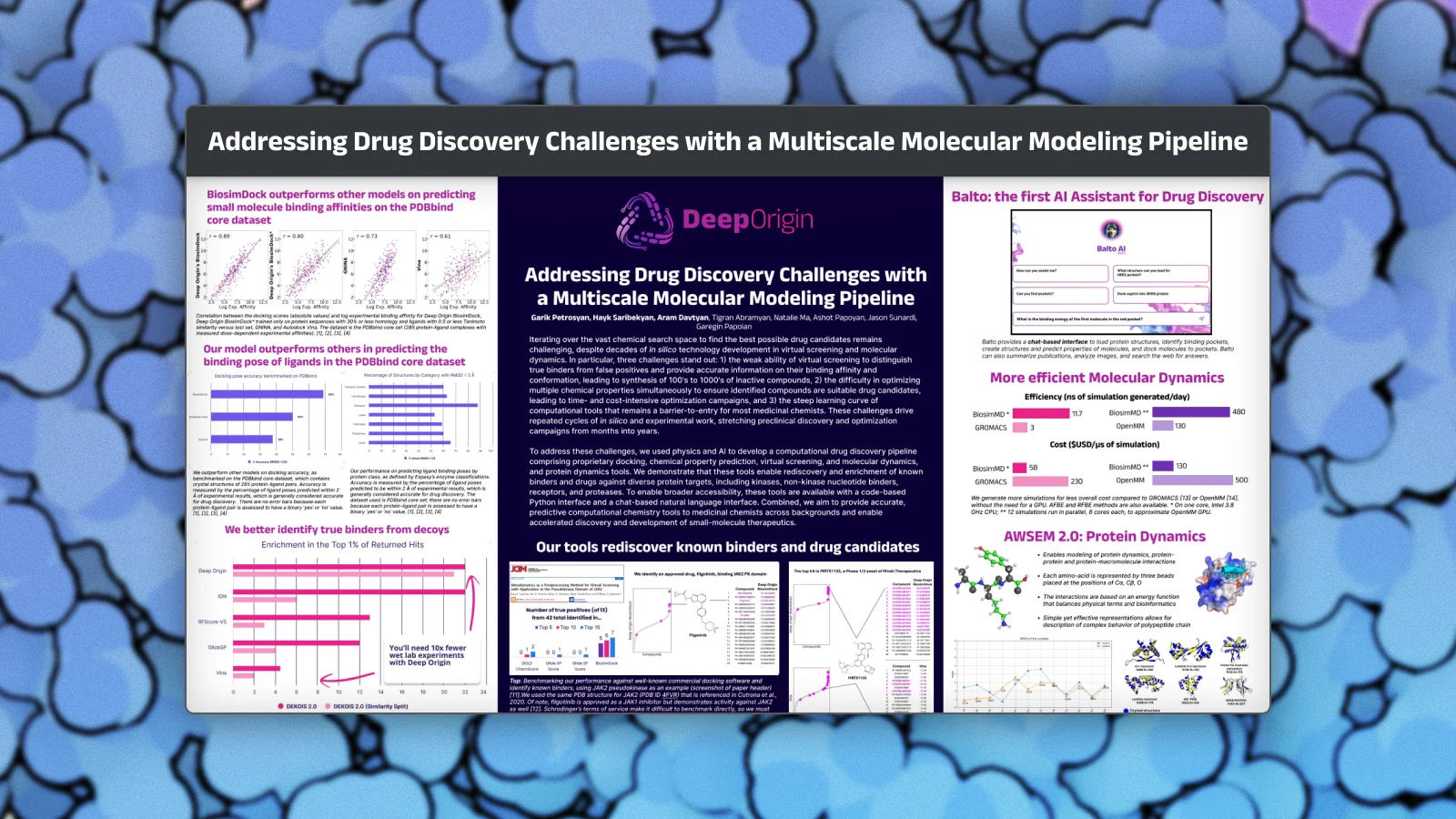
Addressing Drug Discovery Challenges with a Multiscale Molecular Modeling Pipeline
Presented at the American Society of Chemistry’s National Medicinal Chemistry Symposium (ACS NMCS)
Iterating over the vast chemical search space to find the best possible drug candidates remains challenging, despite decades of in silico technology development in virtual screening and molecular dynamics. In particular, three challenges stand out: 1) the weak ability of virtual screening to distinguish true binders from false positives and provide accurate information on their binding affinity and conformation, leading to synthesis of 100’s to 1000’s of inactive compounds, 2) the difficulty in optimizing multiple chemical properties simultaneously to ensure identified compounds are suitable drug candidates, leading to time- and cost-intensive optimization campaigns, and 3) the steep learning curve of computational tools that remains a barrier-to-entry for most medicinal chemists. These challenges drive repeated cycles of in silico and experimental work, stretching preclinical discovery and optimization campaigns from months into years.
To address these challenges, we used physics and AI to develop a computational drug discovery pipeline comprising proprietary docking, chemical property prediction, virtual screening, and molecular dynamics, and protein dynamics tools. We demonstrate that these tools enable rediscovery and enrichment of known binders and drugs against diverse protein targets, including kinases, non-kinase nucleotide binders, receptors, and proteases. To enable broader accessibility, these tools are available with a code-based Python interface and a chat-based natural language interface. Combined, we aim to provide accurate, predictive computational chemistry tools to medicinal chemists across backgrounds and enable accelerated discovery and development of small-molecule therapeutics.


