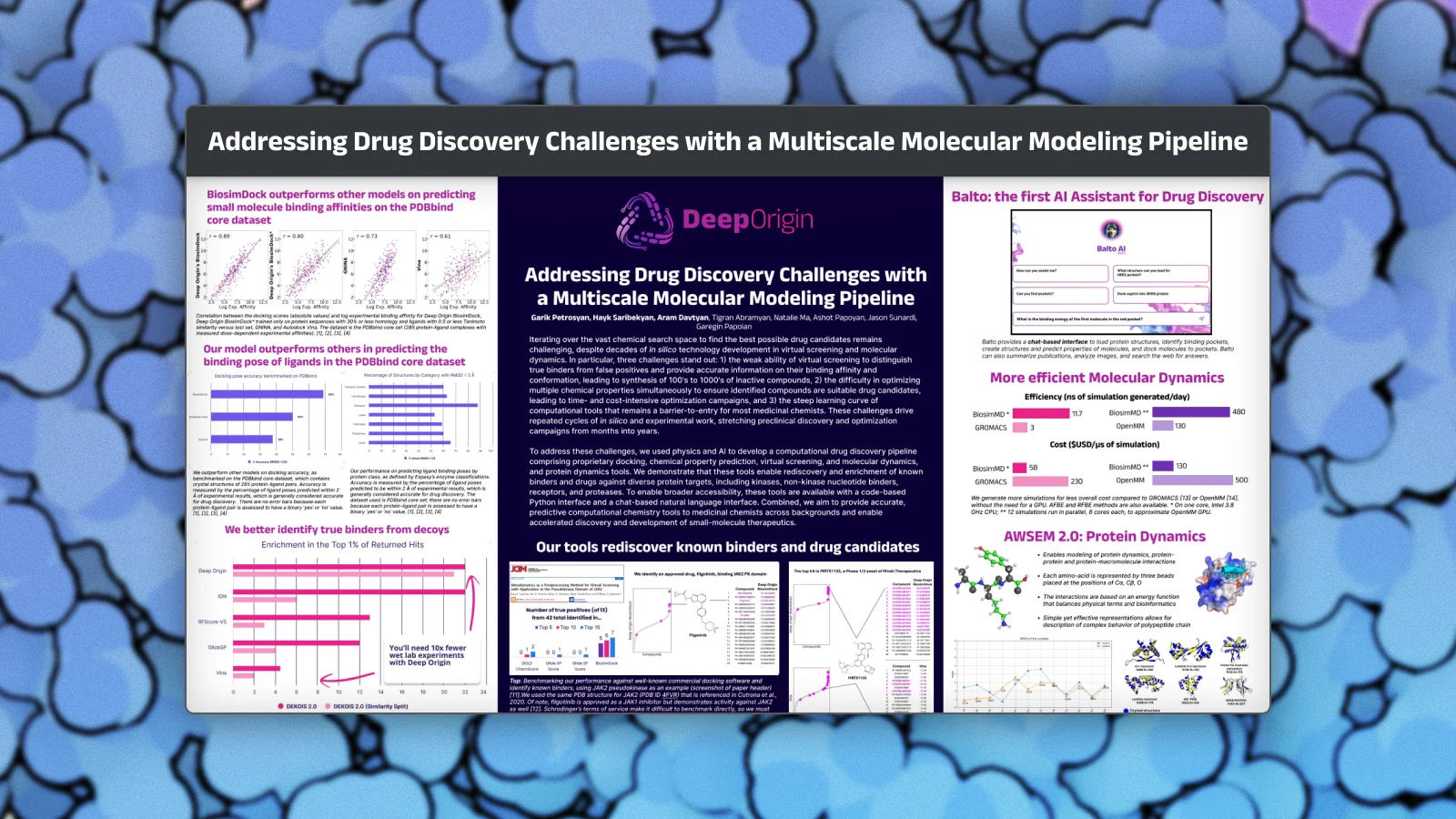
Accelerating Drug Discovery with Physics-Informed Machine Learning

Deep Origin researchers present advances in computational drug discovery combining physics-informed machine learning with large-scale biochemical datasets to improve molecular docking accuracy and binding predictions.
The Challenge
Advances in scale have outpaced improvements in accuracy in virtual screening. This poster demonstrates how physics-informed ML models can achieve substantial gains in pose accuracy, affinity rank-ordering, and early enrichment across retrospective benchmarks.
Technical Focus Areas
- Pose prediction improvements - Enhanced accuracy in predicting molecular binding poses
- Binding-affinity estimation - More reliable predictions of how strongly molecules bind to targets
- Out-of-distribution performance - Strong results on challenging targets including CD73
- End-to-end integration - Seamless pipeline spanning docking, property prediction, virtual screening, reinforcement learning, and molecular dynamics
Delivery Methods
Our physics-informed ML capabilities are available through:
- Python API - Full programmatic access for computational scientists
- Balto AI - Natural-language interface for molecular modeling, making cutting-edge tools accessible to all researchers